Structural biology
Structural biology program follows the premise that physiological processes can be reproduced in vitro once we know all molecular components and understand their properties and interactions. Such understanding requires insight in the molecular mechanisms from the points of view of structural, molecular, and computational biology. It is the atomic 3-dimensional structures of macromolecules in particular that link the information from the sequences of biological polymers with the role of individual residues in biophysical and chemical mechanisms underlying physiological processes.
The main biological theme of the proposed research is the process of adaptive immunity, where organism learns to discriminate between self and non-self proteins as a specific defense against invading microorganisms and their own misbehaved cells. This process begins with antigen presentation. Peptidyl candidates for antigenic presentation are generated by degradation of foreign proteins by endosomal proteases and other hydrolytic enzymes and continue with two level selection process, including binding to the MHC class II molecules and recognition of the formed complexes by the T-cells.
The Structural biology program is a continuation of a decade of our research. Recently, we established a platform for high-throughput, parallelized, and semi-automated protein expression, purification, crystallization, and structure determination. This pipeline enables us to work simultaneously with groups of proteins, performing functional studies of experimental models with increasing complexity. In addition to continuing to study endosomal proteases, we will also study other endosomal enzymes, MHC class II molecules, their targets from the surface of bacterial pathogens Staphylococcus aureus, Clostridium difficile, and Enterococcus faecalis, and the selection process of antigen presentation. We aim to elucidate what makes a protein substrate of a cysteine cathepsin and to develop a method to predict this property. We expect that 3D structure of MHC class II molecules in complex with invariant chain will provide clues that will enable us to interpret in detail the complicated mechanism of invariant chain processing that occurs simultaneously with proteolytic degradation of antigenic proteins. We expect that continuation of our studies of proteins from pathogenic bacteria involved in the structure and remodeling of surface assemblies, such as S-layer, peptidoglycan cell wall, and biofilms, will help us to explore their potential as targets for novel anti-bacterial therapies. To pursue our goals, we will use biochemistry, molecular and cell biology, macromolecular crystallography, electron microscopy, small angle X-ray scattering, organic synthesis, proteomics, and computational biology. Such broad spectrum of technologies can only be applied within an extensive local and international network of collaborators.
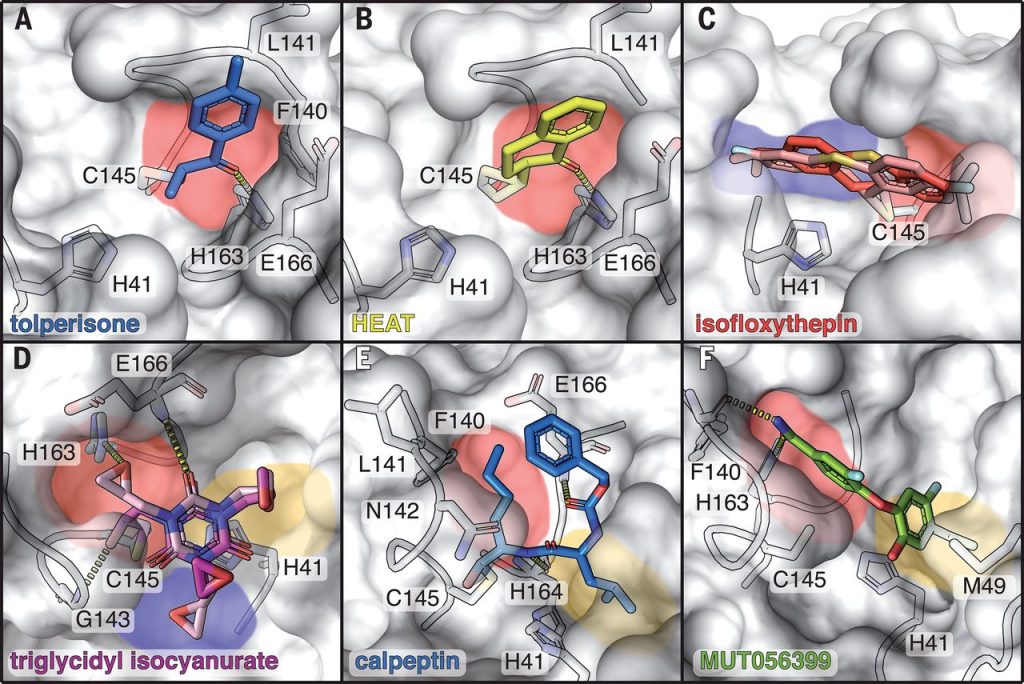
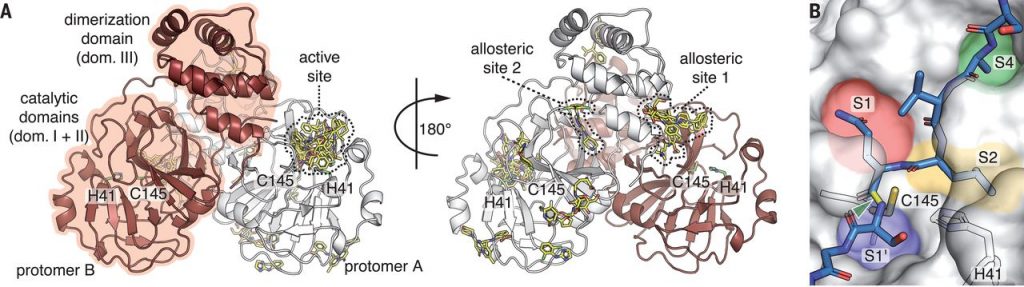
Figure 1: X-ray screening of drug-repurposing libraries reveals compound binding sites distributed across the complete Mpro surface. (A) Schematic drawing of Mpro dimer structure. Protomer A in white, protomer B in red. For clarity, the 29 binding compounds (yellow sticks) are only depicted on one of the two protomers. Catalytic residues H41 and Cys145, active site and two allosteric drug binding sites are highlighted. (B) Close-up view of active site with peptide substrate bound (blue sticks), modelled after SARS-CoV Mpro (PDB 2Q6G). Scissile bond is indicated in yellow and with green arrow. Substrate binding pockets S1’, S1, S2 and S4 are indicated by colors. Sebastian Günther et al. (102 authors), “X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease”, Science, 2021, 372, 6542, 642-646.
Slika 1: Presejalni test knjižnic za ponovno uporabo zdravil, opravljen z rentgensko difrakcijo, razkriva vezavna mesta po celotni površini Mpro. (A) Shematski prikaz strukture dimera Mpro. Protomer A je obarvan belo, protomer B rdeče. Zaradi boljše preglednosti so vezane spojine (29), označene rumeno, prikazane le na enem protomeru. Poudarjeni so katalitična preostanka H41 in Cys145, aktivno mesto in dve alosterični vezavni mesti. (B) Pogled aktivnega mesta od blizu z vezanim peptidnim substratom (označene modro), modeliran s pomočjo strukture Mpro iz SARS-CoV (PDB 2Q6G). Cepitveno mesto je poudarjeno z rumeno in označeno z zeleno puščico. Različne barve ponazarjajo različna vezavna mesta za substrat (S1’, S1, S2 in S4). Sebastian Günther et al. (102 authors), “X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease”, Science, 2021, 372, 6542, 642-646.
Figure 2: Covalent and non-covalent binders in the active site of Mpro. Bound compounds are depicted as colored sticks while the surface of Mpro is shown in grey with selected interacting residues as sticks. Substrate binding pockets are colored as in Figure 1. Hydrogen bonds are depicted by dashed lines. (A) tolperisone, (B) HEAT, (C) isofloxythepin, (D) triglycidyl isocyanurate, (E) calpeptin, (F) MUT056399. Sebastian Günther et al. (102 authors), “X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease”, Science, 2021, 372, 6542, 642-646.
Slika 2: Kovalentno in ne-kovalentno vezane spojine v aktivnem mestu Mpro. Vezane spojine so prikazane z različnimi barvami, površina Mpro je obarvana sivo, prikazani so tudi preostanki, ki so udeleženi v vezavo. Barve, ki označujejo vezavna mesta za substrat, so enake kot na sliki 1. Vodikove vezi so prikazane s črtkano črto. (A) tolperison, (B) HEAT, (C) isofloksitepin, (D) triglicidil isocianurat, (E) kalpeptin, (F) MUT056399. Sebastian Günther et al. (102 authors), “X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease”, Science, 2021, 372, 6542, 642-646.